Refinement of Molecular Descriptors for Diversity Analysis of Chemical Libraries
The search for lead compounds using computational tools (aka, virtual screening) requires chemical libraries. But there are many chemical libraries and it is time-consuming to search all of them. Therefore, a tool is required to decide on the similarity of these libraries and assess in selecting a diverse set of compounds that is representative of all available libraries. Such a tool uses molecular descriptors (such as BCUT) to perform the task. Herein, we refined these descriptors and verified that their performance has improved.
Development of Fragment-based Molecular Descriptors using Subgraph Mining
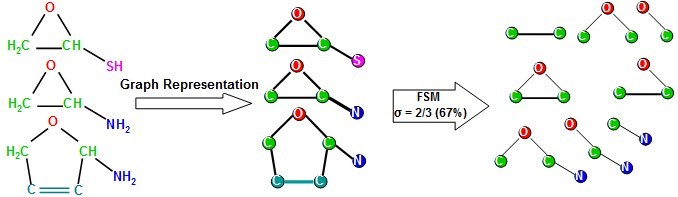
Molecular descriptors are also useful for performing quantitative structure-activity relationship (QSAR) studies as well. In these studies, a regression (or classification) model is generated to relate a set of molecular descriptors to the activity of the molecules in the study. The model can then be used to predict the activity of other molecules in a virtual screening process. Molecular descriptors are of many kinds, the fragment-based ones use counts of chemical fragments to describe molecules. Thus, they afford a mechanistic interpretation of the results in terms of essential pharmacophoric (or toxicophoric) elements responsible for the activity (or toxicity) of molecules. Herein, we developed fragment-based molecular descriptors using frequent subgraph mining (FSM); we used a labeled chemical graph representation of molecules and employed FSM to identify chemical-fragments (subgraphs) that occur in at least a fraction (σ) of all molecules in a dataset, see the following figure. We demonstrated (using variable-selection QSAR modeling) that the identified molecular descriptors afford higher discriminatory ability in classifying compounds compared to other commonly used molecular descriptors.
Development of Topological (2D) Pharmacophore Models using Subgraph Mining
Using the same steps in the previous figure, we can identify the common pharmacophoric groups in a set of active molecules sharing the same pharmacological effect. But this time we employed the classification-based association (CBA) technique, which selects the best combination of the fragments that are able to classify molecules as active or inactive. We demonstrated that the method affords higher discriminatory power in classifying compounds compared to other commonly used methods. Thus, it can be used in virtual screening for lead identification.
Developing Novel Scoring Function (MBI-Score) for Docked Protein-Ligand Complexes
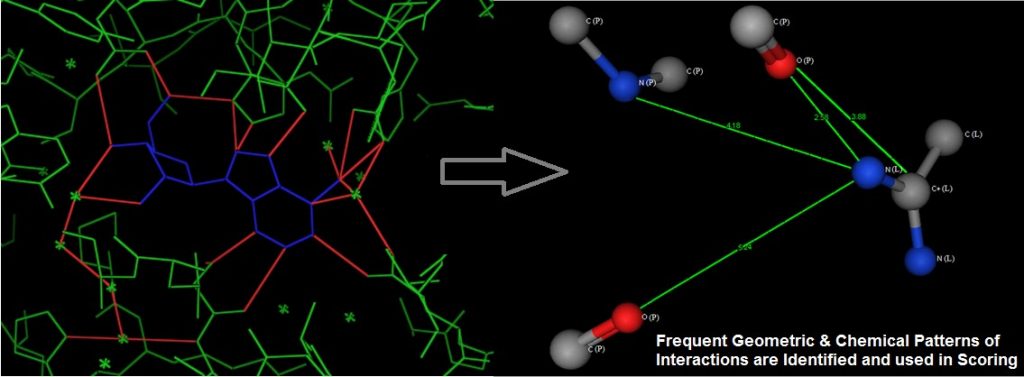
We used graph representation of protein-ligand interfacial atoms in native complexes, and employed subgraph mining to extract patterns of atomic interaction. These are then used to score docked protein-ligand complexes to identify the correct binding mode, see the following figure. The uniqueness of this method is that it takes into account the ‘multi-body’ interaction effect, thus improving accuracy over current methods which use the summation of pair-wise interactions.
Generating “Fragment-based Virtual Library” (FragVLib) by Pocket Similarity Search
FragVLib employs graph representation of interfacial atoms of native protein-ligand complexes and performs pocket similarity search to extract fragments sharing the same binding region. These can then be augmented to form new compounds and tested experimentally.
Formulate an Algorithm for Estimating the Entropy
In the field of computational chemistry, there are two major challenges. First, sampling of all possible conformation of molecules, particularly large ones such as proteins; second, the accuracy of scoring functions that are responsible for calculating the binding affinity between interacting molecules. The reason behind the inaccuracy of scoring functions is mainly due to the lack of the contributions of conformational entropy and solvation energy. To this end, we formulated an algorithm to estimate the entropy using the steepness of the curve of state energy; see the following figure.