Given the 3D crystal structure of such complexes, we use molecular dynamics (MD) simulation to gain perception of the structure and dynamics of the association process, and thus, arrive at a complete mechanistic understanding of how the binding occurs, and what are the changes this binding induces to the conformations which, as a result, triggers the signal transduction process. We also quantify the binding affinity of the association process by estimating the free energy of binding using large-scale MD simulation. The simulation model will be evaluated by its ability to reproduce experimental measures such as binding affinity. Such insight and advancement in this field will improve our understanding of biological processes, and thus, support rational drug design and facilitate the drug discovery process.
Investigating the Molecular and Structural Mechanism Underlying DNA Integration by Transposon-encoded CRISPR-Cas Systems to Support the Development of Highly Accurate and Efficient Gene-Editing Tools
CRISPR-Cas9, a popular genome-editing tool that allows researchers to make double strand breaks to edit parts of the genome by inserting, adding, or changing the specific sequence in the DNA. It is simple, efficient, and customizable which is advantageous for CRISPR-Cas9 over the other gene-editing tools. However, CRISPR-Cas9 might cause unintended double-strand breaks in DNA leading to an off-target effect. There have been many improvements to the CRISPR-Cas system, but up until recently, all trials failed to insert DNA into specific locations without causing the toxic double strand breaks in DNA. Sternberg group at Columbia University (a collaborator) have discovered a new system known as Transposon-encoded CRISPR–Cas (aka, INTEGRATE) that can accurately insert DNA sequences without causing a double-strand break in the target DNA. The INTEGRATE system starts like CRISPR-Cas9 but has a different ending. This newly discovered system is very promising since it is very efficient and very accurate gene-editing tool. Recent experimental results by two groups, Sternberg and Peters, have revealed three critical stages responsible for the on-target RNA-guided DNA transposition by INTEGRATE: recognition of the target DNA, completion of an intermediate (R-loop) state, and activation of the transposition regulator TnsC. However, the atomic level details of the binding pathways, activation and recruitment have not been discovered since it is a challenging experimental task. Here is where the power of Molecular Dynamic (MD) simulation comes into play. To this end, I will utilize MD simulation to shed light on the three key stages and obtain atomic-level details on the conformational changes that take place in the INTEGRATE system. This study will provide new insights into the molecular mechanisms of DNA targeting and improve the engineering of CRISPR-Cas systems with higher on-target accuracy and efficiency.
The following figure describes DNA insertion by INTEGRATE. It starts with assembly of the Cascade-RNA-TniQ complex in a helically twisted “G” shape. Next is recognition of the Protospacer Adjacent Motif (PAM) sequence in the target DNA by Cas8 and formation of a seed-bubble which grow into an R-loop. When R-loop is complete it triggers the activation of TniQ which, in turn, activates TnsC. Finally, TnsC will unwind the target DNA and recruit TnsB and TnsA to perform the DNA transposition process. Transposition of the DNA cargo (pink lines) occurs 49 bp downstream of the DNA target. Complete R-loop formation is one of the transition states used as a checkpoint to ensure that DNA insertion occurs in the correct place “on-target” where there is a full complementarity between RNA and DNA.
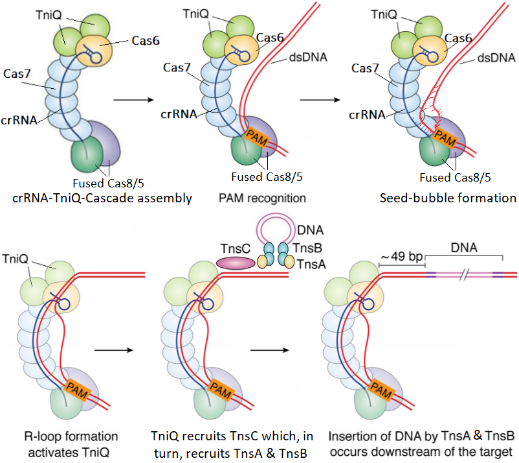
Herein, in collaboration with Sternberg group, we are utilizing the high resolution images of the INTEGRATE system to perform MD simulation studies to provide rigorous mechanistic insight into the structural conformational changes to the INTEGRATE system at three crucial stages: PAM recognition and seed-bubble formation; R-loop stabilization and activation of TniQ; and activation of TnsC and unwinding of DNA.
Investigating the Molecular and Structural Mechanism Underlying Insulin Receptor Activation to Support Drug Discovery Efforts
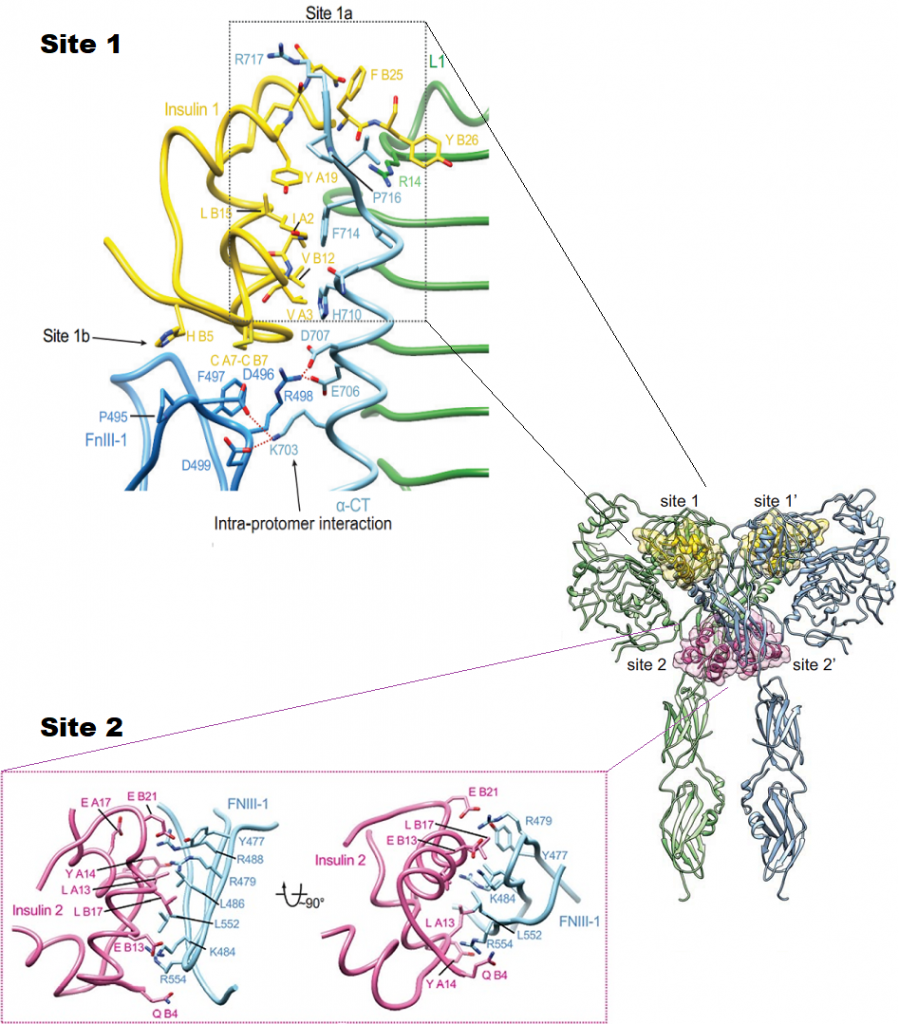
The insulin receptor (IR) has a pivotal role in the regulation of glucose homeostasis, and its dysfunction can result in a range of clinical manifestations, including diabetes mellitus and cancer. Despite deep interest in understanding transmembrane signaling by the insulin receptor (IR), the underlying molecular structural mechanism of IR activation remains elusive. IR is made of two copies of protein chains (homodimer) that come together on the outside of the cell to form the receptor site (ectodomain) that binds to insulin. Each protein chain is connected through the membrane to a tyrosine kinase. In absence of insulin, the chains are held in a constrained position, but when insulin binds, these constraints are released. They first phosphorylate and activate each other, and then phosphorylate other proteins in the signaling network inside the cell. Transmembrane signaling by the IR must involve structural changes within the receptor dimer. Recently, major progress has been made in uncovering the mechanisms of how insulin engages its receptor and modulates downstream signal transduction. A novel kinase triggering mechanism suggests that insulin, after its initial interaction with binding site 1, engages with site 2 between the fibronectin type III(FnIII)- 1 and -2 domains, which changes the conformation of FnIII-3 and eventually translates into structural changes across the membrane. This structural rearrangement of the ectodomain propagates to the transmembrane domains, which are well separated in the inactive conformation but come close together upon insulin binding, facilitating autophosphorylation of the cytoplasmic kinase domains. However, the atomic level details of the binding pathways, structural rearrangements, and allosteric communication between domains have not been discovered since it is a challenging experimental task. Here is where the power of Molecular Dynamic (MD) simulation comes into play. To this end, we will utilize MD simulation to obtain atomic-level details on the conformational changes that take place upon binding of insulin and activation of its receptor. This study will provide new insights into the molecular mechanisms of IR activation and, thus, could lead to future novel drug discovery efforts.